NMR生データを手に入れたら〜本編
このページは、本学部装置のユーザー向けに作製したTopSpinマニュアル(私家版)をWeb化したものを許可を得て公開しています. 測定データを別のパソコンにインストールしたTopSpin(Process Only) で扱う場合の操作を中心に解説しています.図を作成したのはWindows版TopSpinでバージョンは主として4.0.7 、一部は3.6、3.7、2.1です.数字の小さいバージョンのTopSpinで測定したデータを、以降のバージョンのTopSpinで開くことは問題ありません.データを測定した装置はBruker Avance I および Neo, またソフトウエアについてくる見本データです.
本編では最低必要なデータ処理を、導入編ではインストールや設定・基本操作について扱います.応用編に必要に応じて行う処理などを加筆していきます.
コマンド入力でapbk[ENTER]すると自動で位相とベースラインが補正される.
スペクトルを縦方向に拡大して、ベースラインが一続きになっていない所があれば直す.
自動処理では、DEPT135°、二次元のHSQC、NOESYで上下が反転していることがある.
ソフトによらず、位相補正のゼロ次の補正はスペクトル全体に効き、一次の補正は周波数に依存した補正になる.
その後、コマンドbas[ENTER]→Auto-correct baseline using polynominalでベースライン補正をする.
化学シフト合わせを行ったら、その前に(自動で)行われたピークピックをやり直す必要がある.
カーボンの化学シフトを合わせた後、任意のピークで合わせるか、または、カーボンのProcParsタブの中のSR[Hz]を読み取り、DEPTのSRに同じ値を入力する.
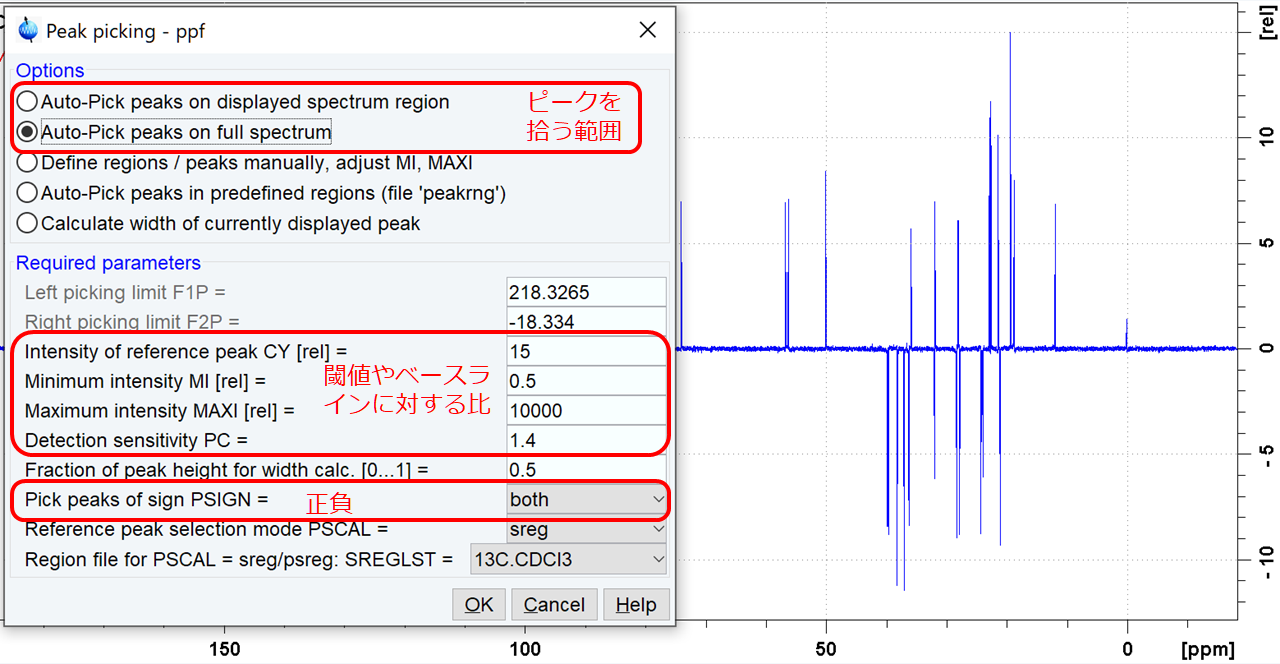
pp[ENTER]して現れるウインドウで感度やスレッショルドを設定し、OKする.PROCPARSタブのPeaksの項目でも同様の設定ができる.
表示させている範囲のピークを拾うか、全体のピークを拾うかを切り替えられる.
CY[rel]に対する最小値のMIと、ノイズに対する感度のPCのいずれか大きい方の基準でピークが拾われる.MAXIを小さくすると大きなピークを拾わないようになる.負のピークも拾いたいとき(DEPT135など)はPSIGNをbothにする.
Analyse→Pick Peaks→Manual-Peak Pickingとしてピークピックモードに入る.
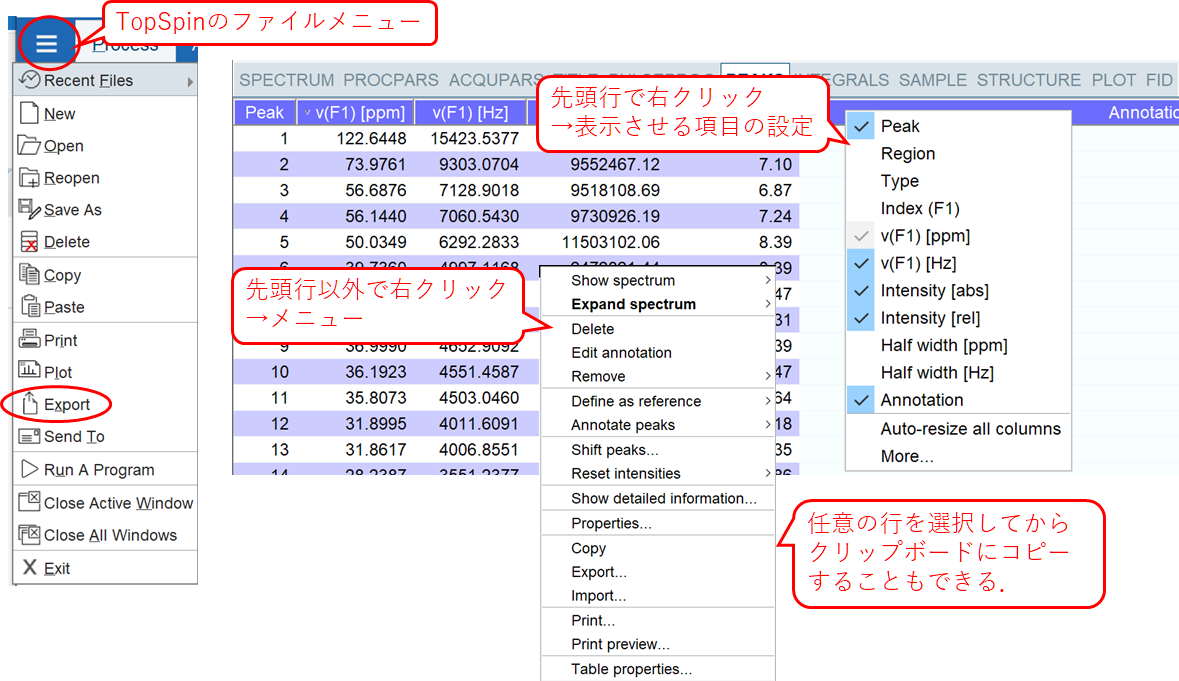
ピークテーブルは,PEAKSタブで表示する.TopSpinメニューバーのファイルメニューのExportから、または、PEAKSタブの表示領域で右クリックしたメニューから、ファイルへの書き出しなどを行う.CSVで保存すればスプレッドシートで開ける.
すでに(自動で)行われた積分の変更も.
Analyse→Integral→Manual-integrate、またはAnalyse→Integral▼をクリック、または.int[ENTER]として積分モードに入る.
Spectrumタブで積分曲線や積分比の表示を変えるには、スペクトル表示領域で右クリック→Spectra Display Preferenceを設定する.
積分結果はINTEGRALSタブで表示し、Peaksタブと同様にexport等を行う.
測定したPC以外でデータを扱う際、Spectrumタブでは良いように見えても、Plotタブでは表示されないことがある.このときも、以下のa,bいずれかの手順で指定し、Spectrumタブからコマンドplot[ENTER]するとなおる.
1D参考スペクトルを縦拡大したいときは、スペクトルの斜め上にある□をクリックして■の状態にしてから、1Dスペクトルの要領で行う. F1、F2とも1Dスペクトルが選択されていない(□)場合は、その操作は2Dスペクトルに対して作用する.Plotタブでもほぼ同様にできる.
化学シフトを合わせたいピークを拡大しておいてから、Process→Calib Axis すると、カーソルが表れる.
ピーク中央でクリックすると化学シフトを入力するウインドウが表れるので、数値(一次元スペクトルで読み取る)を入力してOKする.
化学シフト合わせに関係するパラメータは、ProcParsタブの中のoffset[ppm]、SR[Hz]であり、一方を変えると他方も変変化し、化学シフト合わせの状態も変わる.
一次元の化学シフトを合わせた後のSRの値を二次元スペクトルの同じ核の軸に設定すれば二次元スペクトルの化学シフトが合わせられる. また、DEPTスペクトルに、13C一次元測定で溶媒ピークで化学シフトを合わせたのちのSR値を設定すると化学シフトが合う.
下図は青が正,赤が負である.
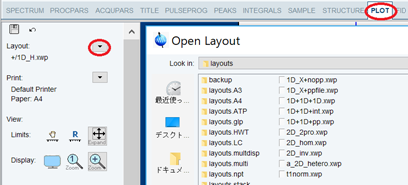
PLOTタブをクリックする.
Layoutの右の をクリックしてレイアウトファイルを選択する.レイアウトファイルはカスタマイズして保存しておくことも可能.PDFや紙で保存する場合、かならずパラメータをつける.二次元で縦方向にも1Dスペクトルを貼り付けるには2D_2pro.xwpを元にすると良い.
スペクトルをクリックすると緑色の■が現れ,左のメニューもスペクトル編集用に変化する.この状態でのスペクトルの拡大は,SPECTRUMタブと同様に行える.2Dスペクトルで1D部分をクリックするとその部分が編集可能となる.選択解除はSpectrumタブとは異なり、別の部分を選択する.
メニューの青文字をクリックすると,サブメニューが開き,書き出し範囲などを設定する.
変更が思った通りに反映されない場合、Spectrumタブに戻り、コマンドplot[ENTER]する.
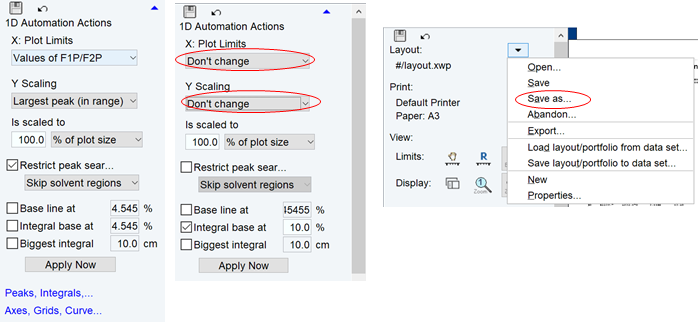
Printの右の をクリックして印刷するか、TopSpinのメニューバーの Plotタブは開くたびに表示範囲がリセットされる設定(下図左)になっていることがある.これは多くの似たサンプルの条件をそろえて自動で書き出しするときには便利な設定で、範囲のスペクトル左端と右端のppm設定(FP1, FP2)はProcParsタブの値に従う.
この設定が不便な場合、下図中央のようにXやYをDon’t changeにして、Apply Nowしてから、レイアウトファイルをSave asし、これをLayoutに設定すると良い.それでもタブを開くと表示はリセットされやすいので、一気に描き上げる方がよい.
(参考)ファイルメニューからのいろいろな出力
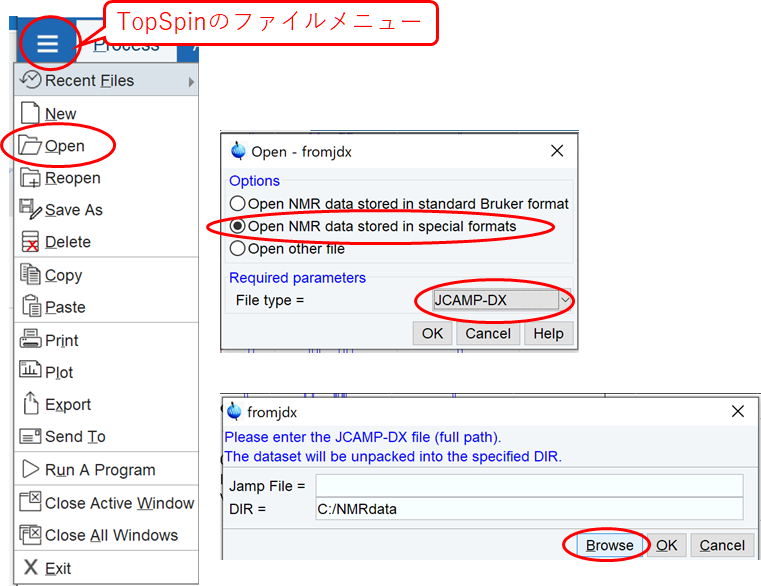
JEOL EX-270で保存したスペクトルデータ(*.als)は、EX270のExcaliburでデータをJCAMP形式に変換しておく必要がある.
FIDデータ(*.gxd) やそれ以降のJEOL装置のデータ(*.nmf, *.bin)はOpenからFile Type=JNMRとして開く.
位相補正
![]() となり位相補正モードに入っていることを示している.
となり位相補正モードに入っていることを示している.
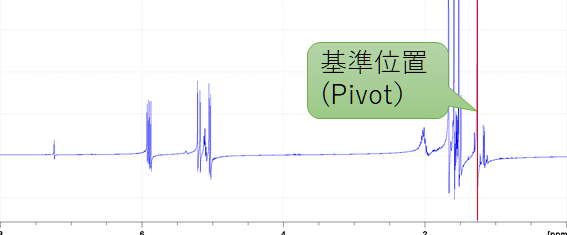
![]() の上でマウスをドラッグしゼロ次補正をする.
の上でマウスをドラッグしゼロ次補正をする.
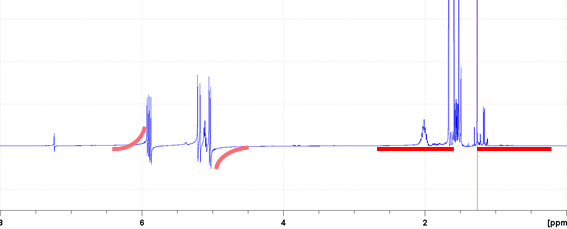
![]() の上でマウスをドラッグして,ここから遠い位置のピークの左右のベースラインに着目して一次補正する.
の上でマウスをドラッグして,ここから遠い位置のピークの左右のベースラインに着目して一次補正する.
![]() ,ゼロ次を指定の角度変えるには90や180(上下反転) ,マウスの感度を変えるには
,ゼロ次を指定の角度変えるには90や180(上下反転) ,マウスの感度を変えるには![]() アイコンを使う.
アイコンを使う.
![]() をクリックして位相補正を保存してルーチンから抜ける.
をクリックして位相補正を保存してルーチンから抜ける.
化学シフト合わせ
DEPTの化学シフト合わせ
ピークを拾う
拾えなかったピークを拾うには
![]() がピークピックモードであることを示している.
以下はそれぞれのアイコンをクリックして背景色が濃くなればその操作ができ,もう一度クリックすればマウスの動作はスペクトルを拡大等になる.
がピークピックモードであることを示している.
以下はそれぞれのアイコンをクリックして背景色が濃くなればその操作ができ,もう一度クリックすればマウスの動作はスペクトルを拡大等になる.
![]() アイコンをクリック→マウスドラッグでピークを拾う.このときの閾値と感度は上記で設定した通りになる.それ以下のピークを拾うには、
アイコンをクリック→マウスドラッグでピークを拾う.このときの閾値と感度は上記で設定した通りになる.それ以下のピークを拾うには、![]() →右クリック→Add Peak To Listするとそのカーソル位置が拾われる.すべて,または一部のピークピックを取り消すときは,
→右クリック→Add Peak To Listするとそのカーソル位置が拾われる.すべて,または一部のピークピックを取り消すときは, ![]() を使う.
を使う.
![]() をクリックして保存して抜ける.
をクリックして保存して抜ける.
積分する
![]() が積分モードであることを示している.それぞれのアイコンをクリックして背景色が濃くなればその操作ができ,もう一度クリックしてどこも灰色が濃くなければ、マウスの動作はスペクトルの拡大等になる.
が積分モードであることを示している.それぞれのアイコンをクリックして背景色が濃くなればその操作ができ,もう一度クリックしてどこも灰色が濃くなければ、マウスの動作はスペクトルの拡大等になる.
![]() ドラッグして積分する.
ドラッグして積分する.![]() 積分曲線を切る.積分曲線のベースライン位置や縦方向の拡大を変更するには
積分曲線を切る.積分曲線のベースライン位置や縦方向の拡大を変更するには![]() を使う.
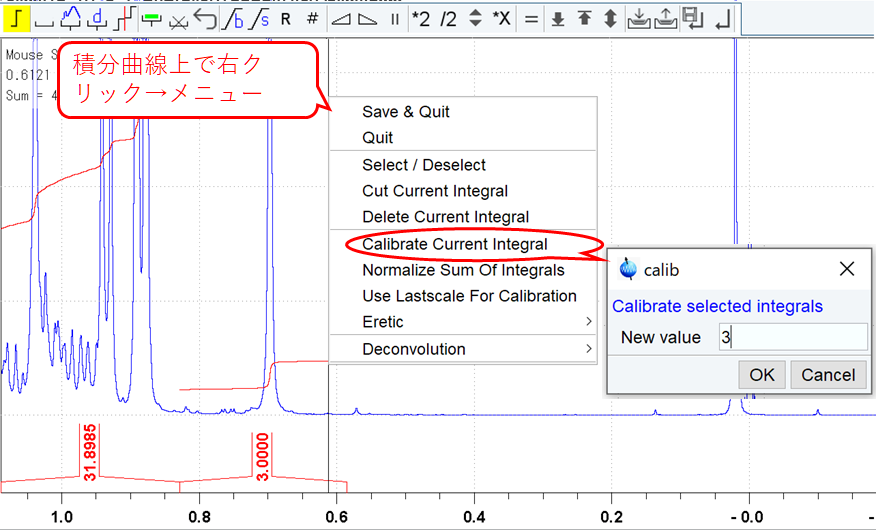
積分を消したいとき,積分比を設定したいときは,積分曲線上で右クリックして現れるメニューで操作する.
保存して抜ける場合は、
を使う.
積分を消したいとき,積分比を設定したいときは,積分曲線上で右クリックして現れるメニューで操作する.
保存して抜ける場合は、![]() をクリックする.
をクリックする.
二次元スペクトル
二次元の上、左に1Dスペクトルを表示することができる
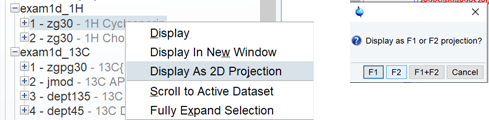
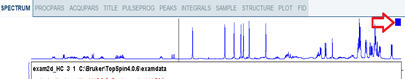
二次元の化学シフトあわせ
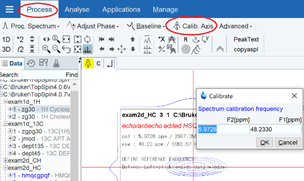
化学シフト合わせの別法
二次元の位相補正(HSQC, NOESY)

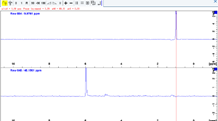
![]() で保存してrow/colの補正モードから抜ける.必要なら縦横両方向補正する.
で保存してrow/colの補正モードから抜ける.必要なら縦横両方向補正する.
二次元位相補正できないとき
※ Cannot phase rows, imaginary part 2ir is missing などと表示される場合は、位相補正モードからいったん抜けて、Process→Advanced→Special Transform→Hilbert in F2 (xht2) を選択する.または、位相補正モードからいったん抜けて、コマンドxht2[ENTER]する.
書き出し(PDF・紙)
 必要ならPrinterを設定する.
必要ならPrinterを設定する.
![]() (File)からExportを選択、保存したいフォルダまで移動して、Filenameに 保存する名前を拡張子つきでと入力しOKする.保存する形式は、拡張子で判定される.
(File)からExportを選択、保存したいフォルダまで移動して、Filenameに 保存する名前を拡張子つきでと入力しOKする.保存する形式は、拡張子で判定される.
PlotタブのAutomation actions

他機種のデータを開くには
*.alsをJCAMP形式に変換する
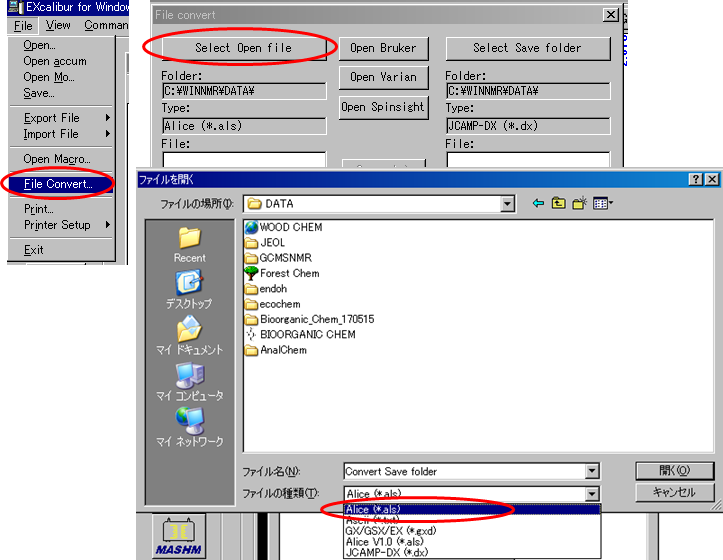
TopSpinでJCAMP形式のデータを開く